
People
Johannes Eller (Alumnus)
Elmar Sauer (Alumnus)
The group of S. Majid Hassanizadeh and Amir Raoof are expected to continue and intensify their scientific work in the framework of SFB 1313. Research Project A01 clarifies thermodynamics phenomena of porous materials in using DFT as a well-founded diffuse interface approach with average molecular resolution. In cooperation with Research Project A02 hydrophilic and hydrophobic materials, but also changes in wettability will be studied. These aspects are also particularly relevant on the small scale and will be studied in cooperation with the group of Majid Hassanizadeh and Amir Raoof at Utrecht University. Joint investigations of free flow/porous-media flow have started.
Publications
Preprints
- Bursik, B., Stierle, R., Oukili, H., Schneider, M., Bauer, G., & Gross, J. (2025). Modelling Interfacial Dynamics Using Hydrodynamic Density Functional Theory: Dynamic Contact Angles and the Role of Local Viscosity. In arXiv preprint arXiv:2504.03032. https://doi.org/10.48550/arXiv.2504.03032
Journal Articles
- Bursik, B., Eller, J., & Gross, J. (2024). Predicting Solvation Free Energies from the Minnesota Solvation Database Using Classical Density Functional Theory Based on the PC-SAFT Equation of State. The Journal of Physical Chemistry B, 128, Article 15. https://doi.org/10.1021/acs.jpcb.3c07447
- Bursik, B., Stierle, R., Schlaich, A., Rehner, P., & Gross, J. (2024). Viscosities of inhomogeneous systems from generalized entropy scaling. Physics of Fluids, 36, Article 4. https://doi.org/10.1063/5.0189902
- Stierle, R., Bauer, G., Thiele, N., Bursik, B., Rehner, P., & Gross, J. (2024). Classical density functional theory in three dimensions with GPU-accelerated automatic differentiation: Computational performance analysis using the example of adsorption in covalent-organic frameworks. Chemical Engineering Science, 298, 120380. https://doi.org/10.1016/j.ces.2024.120380
- van Westen, T., Hammer, M., Hafskjold, B., Aasen, A., Gross, J., & Wilhelmsen, Ø. (2022). Perturbation theories for fluids with short-ranged attractive forces: A case study of the Lennard-Jones spline fluid. The Journal of Chemical Physics, 156, Article 10. https://doi.org/10.1063/5.0082690
- Eller, J., Matzerath, T., van Westen, T., & Gross, J. (2021). Predicting solvation free energies in non-polar solvents using classical density functional theory based on the PC-SAFT equation of state. The Journal of Chemical Physics, 154, Article 24. https://doi.org/10.1063/5.0051201
- Stierle, R., & Gross, J. (2021). Hydrodynamic density functional theory for mixtures from a variational principle and its application to droplet coalescence. The Journal of Chemical Physics, 155, Article 13. https://doi.org/10.1063/5.0060088
- Stierle, R., Sauer, E., Eller, J., Theiss, M., Rehner, P., Ackermann, P., & Gross, J. (2020). Guide to efficient solution of PC-SAFT classical Density Functional Theory in various Coordinate Systems using fast Fourier and similar Transforms. Fluid Phase Equilibria, 504, 112306. https://doi.org/10.1016/j.fluid.2019.112306
- Sauer, E., Terzis, A., Theiss, M., Weigand, B., & Gross, J. (2018). Prediction of Contact Angles and Density Profiles of Sessile Droplets Using Classical Density Functional Theory Based on the PCP-SAFT Equation of State. Langmuir, 34, Article 42. https://doi.org/10.1021/acs.langmuir.8b01985
Published data sets
- Bursik, B., Eller, J., & Gross, J. (2024). Supporting Information: Notebooks, Solute Configurations and Solvation Free Energy Data [DaRUS]. https://doi.org/10.18419/DARUS-3756
- Bursik, B., Stierle, R., Schlaich, A., Rehner, P., & Gross, J. (2024). Additional Material: Viscosities of Inhomogeneous Systems from Generalized Entropy Scaling [DaRUS]. https://doi.org/10.18419/DARUS-3769
Research
About the Project
Project A01 uses molecular modeling of solid-fluid interfaces to understand and predict their effect on fluids porous systems. Density functional theory (DFT) allows to predict interfacial properties in equilibrium, such as adsorption isotherms and contact angles. By developing a dynamic approach based on DFT, wetting can be studied on the molecular scale, e.g. the appearance of dynamic contact angles. The project demonstrates that detailed molecular approaches for the viscosity at solid-fluid interfaces are required to obtain accurate results.
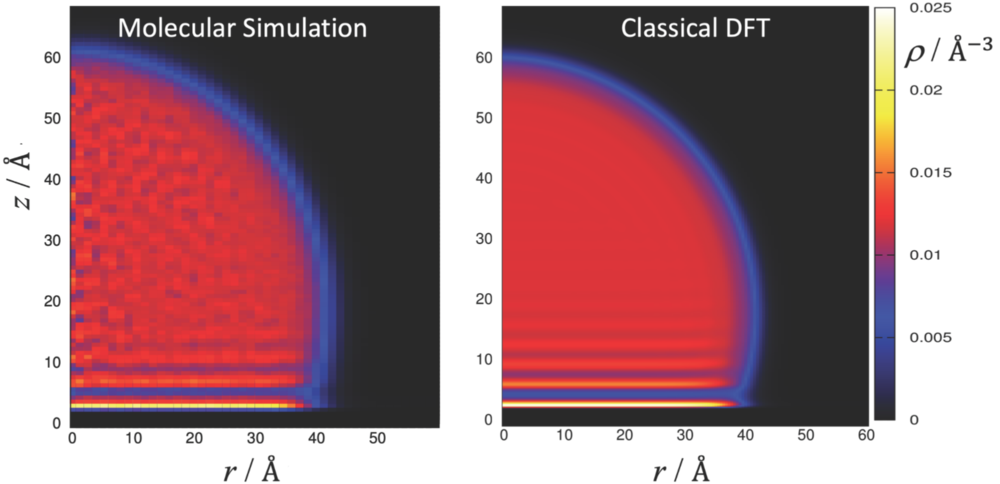
Results
Funding Period 1
In FP1, we developed models and methods to describe fluids and fluid mixtures at solid-fluid interfaces. Using density functional theory (DFT) with a Helmholtz energy functional based on the PC-SAFT model, we accurately predicted adsorption processes and static contact angles. DFT results for pure components and mixtures showed very good agreement with molecular simulations using the same atomistic force field. Contact angle predictions were validated against both simulation and experimental data for pure substances. We also investigated the impact of molecular-scale roughness and chemical heterogeneity on adsorption isotherms, demonstrating that these effects can be effectively averaged into one-dimensional pore–fluid interaction potentials. The work provided predictive methods for equilibrium properties of real substances at interfaces relevant to porous media.
Funding Period 2
In the current (second) funding phase, we continued to study equilibrium systems and we extended our models to dynamic processes. We applied our DFT approach to equilibrium contact angles of macroscopic droplets for a wide range of pure substances and we extended the approach to mixtures. We worked together with project Z02 and compared theoretical results to experimental contact angles. We further demonstrated that our DFT model is applicable to the calculation of solvation free energies.
To describe dynamic processes at the molecular scale, we developed a hydrodynamic density functional theory (DFT) model that combines DFT functionals with component and momentum balance equations. The software for the calculation of the DFT functionals was coupled with the open-source simulator Dumux in a collaboration with projects A02 and INF. A key requirement for the model is the accurate description of local shear viscosity, particularly near solid surfaces. To address this, we collaborated with project C01 and extended the entropy scaling formalism—originally developed for bulk fluids—to solid–fluid interfaces.

Together with project A02 we applied this dynamic approach to study contact angles and the motion of nanoscale droplets on solid surfaces and found good agreement between predictions and non-equilibrium molecular dynamics (NEMD) simulations. Comparison with a simpler, bulk-like viscosity models clearly highlights the importance of using detailed, molecular approaches for the viscosity.

{kind=link}
Future Work
In FP3, we aim to advance the hydrodynamic DFT model to account for non-isothermal processes by incorporating an energy balance alongside component and momentum balances. The non-isothermal hydrodynamic DFT model will predict density, velocity, and temperature fields in dynamic systems, particularly at interfaces where thermal effects are crucial. This project brings a thermodynamically consistent and predictive (molecular-scale) description of interfaces to a continuum-scale model. To determine the required thermal conductivities, we extend the entropy scaling approach, developed in FP2, to thermal conductivities at interfaces and near solid surfaces in collaboration with project C01.
A collaboration with project A02 will focus on the molecular-scale study of droplet behavior on chemically heterogeneous and rough surfaces. Additionally, we will work on evaporation processes in the transition from the porous medium to the free-flow domain and we plan to investigate the Marangoni effect using azeotropic mixtures, both in collaboration with Z02. With C02, we will apply our model to periodic homogenization, and in cooperation with B01, we will study salt formation and convective flow in small-scale pore systems. Together with A06 we are predicting DFT-based interfacial properties from contact angle measurements of SLM-fabricated structures.
We will collaborate with project Maartje Boon by developing predictive models for salt solubility in aqueous systems with dissolved gases, along with transport coefficients. We will further analyze dynamic processes like Ostwald ripening and their interplay with salt formation within the salt vision topic.
International Collaboration
Norwegian University of Science and Technology, Norway
We collaborated with Prof. Øivind Wilhelmsen and developed a DFT approach for fluid mixtures of hydrogen, helium, deuterium and neon, which are strongly affected by quantum effects at low temperatures. This approach can be used to study confined fluids and assess the performance of porous materials for hydrogen storage and transport.
For further information please contact

Joachim Groß
Prof. Dr.-Ing.Project Leader, Research Project A01